Memory and learning - using mouse to model neurobiological and behavioural aspects of Down syndrome and assess pharmacotherapeutics
Mouse models are a standard tool in the study of many human diseases, providing insights into the normal functions of a gene, how these are altered in disease and how they contribute to a disease process, as well as information on drug action, efficacy and side effects. Our knowledge of human genes, their genetics, functions, interactions and biochemistry, has dramatically improved over the last few years. Recently, several different drugs have been shown to rescue learning and memory deficits in a major mouse model of Down syndrome. Here, we first review the challenges inherent in using mouse models in Down syndrome research and then describe the successful molecular/genetic interventions that are cause for cautious optimism. We then predict critical molecular abnormalities that can be tested for relevance to learning and memory and that are potential targets of existing pharmacotherapeutics.
Down Syndrome Research and Practice
Introduction
Mouse models are a standard tool in the study of many human diseases, in basic research and in preclinical assessment of potential therapeutics. Their use, in molecular, cellular and behavioural experiments, can provide insight into the normal functions of a gene, how these are altered in disease and how they contribute to a disease process, as well as information on drug action, efficacy and side effects. Down syndrome has long been regarded by many in both basic and clinical research as too complex a challenge for effective pharmacological intervention. This attitude, however, should change now that several different drugs have been shown to rescue learning and memory deficits in a major mouse model of Down syndrome. The challenges in Down syndrome have not become simpler, rather our knowledge of human genes, their genetics, functions, interactions and biochemistry, has dramatically improved over the last few years. Here, we first review the challenges inherent in using mouse models in Down syndrome research and then describe the successful molecular/genetic interventions that are cause for cautious optimism. Lastly, we predict critical molecular abnormalities that can be tested for relevance to learning and memory and that are potential targets of existing pharmacotherapeutics.
Challenges in modelling Down syndrome in mouse
i) Gene numbers and distribution
Human chromosome 21 encodes over 500 genes [1] . Current knowledge of the functions of these genes is quite limited; we understand something about the function of only one third and nothing at all about the remaining two thirds [2,3] . Because we have no basis for eliminating any genes as candidates for making some contribution to the features of Down syndrome, we are left with a large and complex set of genes to be considered. The basic molecular hypothesis in Down syndrome is that the extra copy of chromosome 21 will result in a 50% increase in the levels of RNA produced from each gene present in three copies. Experiments, with human derived samples and mouse models, have largely verified the prediction of increased RNA [4-8] . Again, data are limited, and they do not allow any significant reduction in the number of genes that need to be considered in a thorough and comprehensive analysis.
ii) Mice are not little people in fur coats
Modelling any human disease in animals is always accompanied by limitations and cautions. Because of the number of genes involved, Down syndrome presents unique as well as common difficulties.
First, when we look at the locations of the best studied of human chromosome 21 genes, we find that they are located on three different mouse chromosomes. Of a set of 170 genes, 112 are on located on mouse chromosome 16, 19 on mouse chromosome 17 and 39 on mouse chromosome 10 (see Figure 1). Thus, creating a mouse that contains three copies of all genes corresponding to those on chromosome 21 is not trivial. In fact, it has not been accomplished, and technical, and possibly biological, difficulties may make it impossible. Given that the largest number of genes map to mouse chromosome 16, these genes have been the focus of model construction. Figure 1 illustrates the current models and the proportion of the 112 genes they contain. The most thoroughly studied of these is the Ts65Dn, because it has been available the longest [9] .
Second, when the genomic DNA of mouse is analysed in detail for gene content, we find significant differences from human chromosome 21. While approximately 540 genes have been identified on human chromosome 21, in the corresponding regions of mouse DNA, only 430 were identified [1] . Furthermore, only 200 human genes are sufficiently similar to mouse to be confident that they have the same functions. Thus, based on current knowledge and with cautious interpretations, we must assume that approximately 300 chromosome 21 genes may have human-specific functions and approximately 200 genes may have mouse-specific functions. The take home message is that, even if we produce a mouse that carries three copies of the appropriate segments of chromosomes16, 17 and 10, it will lack some human genes and carry other mouse genes. While this is a nuisance, it is logically not surprising; it is reasonable to expect the differences between human and mouse are a result of differences at the DNA level.
iii) Assessing learning and memory in mice
Much of human cognitive evaluation relies on language. While this is an obvious limitation in the use of mouse models, there are other correlates of learning and memory that have parallels in rodents and that can be used for evaluation of Down syndrome-relevant deficits and drug efficacies. Most commonly in Down syndrome research these have involved tests of hippocampal-based learning, in particular tests of spatial learning and memory, such as the Morris Water Maze, Contextual Fear Conditioning and Novel Object Recognition. These are important tests because the best assessments of Down syndrome deficits implicate deficits in hippocampal function [14,15] . Notably, the Ts65Dn mice display deficits in each of these tasks [10, 16] .
We also need tests with sensitivity and resolution to discriminate levels of impairment among genetically different mouse models and tests that assay impairment of other specific brain regions that correlate with Down syndrome deficits. We also require uniform application of behavioural testing: reliable comparisons among laboratories of the deficits in different mouse models and of results of therapeutic testing require the use of the same testing protocols. Such criteria have yet to be established within the research community.
Thus, the challenges to use of mouse models in Down syndrome research are the large number of genes on human chromosome 21, the differences in gene content and distribution within mouse chromosomes, and the valid representation and interpretation of complex Down syndrome-relevant learning and memory features in mouse.
Success, and cautions, in rescuing behavioural/neurobiological abnormalities in mouse models
Table 1 lists recent success stories with reversing Down syndrome-like features in Ts65Dn mice. Treatments include three pharmacological reagents: memantine, pentylenetetrazole (PTZ) and fluoxetine (Prozac); two biological reagents: nerve growth factor (NGF) and Sonic Hedgehog (Shh); and one genetic manipulation using the chromosome 21gene APP. The results with memantine and PTZ are particularly exciting because they demonstrate the rescue of learning/memory deficits as measured by three tests of hippocampal function, the Morris Water Maze, Contextual Fear Conditioning and Novel Object Recognition. Other interventions reversed abnormalities seen at the cellular level, e.g. numbers of cells (fluoxetine, Shh) or premature loss of function (NGF, APP), and their effects on learning and memory have yet to be assayed. Table 1 also lists some advantages of each treatment and some significant limitations of the current knowledge.
| Treatment | Target | Phenotypic feature | Outcome | Reference | Advantages | Limitations |
|---|---|---|---|---|---|---|
| Memantine | NMDAR | Hippocampal-based learning deficit (CFC) | Rescue of CFC deficit | Costa et al. [17] | Approved for use in AD | Not tested in children |
| Pentylenetetrazole (PTZ) | GABRA | Hippocampal-based learning deficits (NOR; MWM) | Rescue of NOR, MWM deficits | Fernandez et al. [16] ; Rueda et al. [18] | Effects lasted 3 months after last drug exposure | Not approved for human use; can induce seizures |
| Fluoxetine (Prozac) | Serotonin | Impaired neurogenesis (hippocampus) | Improved neurogenesis | Clark et al. [19] | Approved for human use (mood disorders) | Not tested in children; effects on learning/memory not tested |
| Nerve growth factor (NGF) | TrkA, p75 | Neurodegeneration | Rescue of cellular abnormality | Cooper et al. [20] | - | Effects on learning/memory not tested; no drug available |
| Activator of sonic hedgehog (SHH) | SHH | Cellular abnormalities in the cerebellum | Rescue of cellular abnormality | Roper et al. [21] | - | Effects on learning/memory not tested; increased cancer risk |
| Genetic reduction of APP (chr21 gene) | APP (chr21) | Failed NGF transport, neurodegeneration) | Partial rescue of cellular abnormality | Cataldo et al. [22] ; Salehi et al. [23] | - | Effects on learning/memory not tested; no drug available |
| Tests of hippocampal-based learning: CFC: Contextual Fear Conditioning; NOR, Novel Object Recognition; MWM, Morris Water Maze. AD, Alzheimer's Disease | ||||||
Table 1 | Successful interventions using Ts65Dn mice: pharmacological, biochemical and genetic
A critical point to note in Table 1 is that, with the exception of the APP gene, none of the targets of intervention is a chromosome 21 gene. Instead, different types of information led to the choices of interventions. Memantine and fluoxetine were chosen based on their effects directly on chromosome 21 proteins or indirectly on targets of chromosome 21 proteins. As shown in Figure 2, multiple chromosome 21 proteins, including APP, directly and indirectly impact the activity of the non-chromosome 21 protein complexes, calcineurin (CaN) and the NMDA receptor, NMDAR. Both CaN and NMDAR play critical roles in normal neurological function, including learning and memory. NMDAR is predicted to be over active in Down syndrome due to the combined effects of inhibition of CaN and direct effects of additional chromosome 21 proteins. Memantine antagonises the hyperactivity of NMDAR and thus was predicted, correctly, to reverse effects of at least some chromosome 21 proteins [17] .
Use of fluoxetine (Prozac), which targets the serotonin system, is also based on a mechanistically sound prediction connected to chromosome 21 proteins. Neurogenesis, production of new brain cells from neural precursors, is a normal ongoing process in the adult hippocampus, and is believed to be required for normal hippocampal function. It has been shown to be impaired in the Down syndrome mouse model Ts65Dn [19, 24] . The implication is that impaired neurogenesis will result in impaired hippocampal function. Overexpression of the chromosome 21protein GIRK2 is predicted to impact the serotonin receptor as shown in Figure 3 and as a result to impair neurogenesis. Figure 3 shows that perturbation of the serotonin pathway in Down syndrome is complicated by interactions with additional chromosome 21 proteins.
The remaining treatments, PTZ, NGF and Shh, lack demonstrated connections to chromosome 21 proteins and were chosen instead because of their predicted effects on cellular/functional abnormalities observed in the Ts65Dn mice. However, PTZ effects on GABRA may be mediated through CaN [28] and therefore also have an indirect association with multiple chromosome 21 proteins as shown in Figure 2.
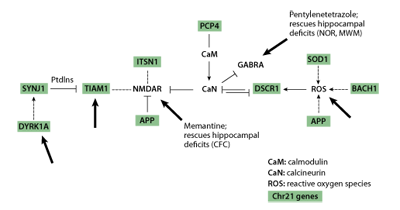
Figure 2 | Chromosome 21genes directly and indirectly impact calcineurin (CaN) and the NMDA receptor (NMDAR). Chromosome 21genes are shown shaded. Large arrows indicate targets of known pharmacological reagents.
The results in Table 1 emphasise several important issues. First and most positively, non-chromosome 21 proteins can be successful, and may be preferable, targets for therapeutics when they can 'integrate' the effects of multiple chromosome 21 proteins. This is clearly illustrated in Figures 2 and 3. The reason for optimism here lies in the possibility that one or a small number of therapeutics may be combined to ameliorate the effects of many critical chromosome 21 proteins, thus making the multi-gene problem more tractable. Second, while our knowledge of the functions of chromosome 21 genes is incomplete, we can still form testable hypotheses regarding potential therapeutics and examine their efficacy in mouse. Third, there are specific very positive features of some of the interventions in Table 1. Memantine and fluoxetine are already approved for human use (in Alzheimer's Disease and depression, respectively); such approval greatly facilitates clinical trials and review processes when and if extension of their use to Down syndrome is considered. It is also encouraging that the effects of PTZ lasted for at least three months following the last drug treatment.
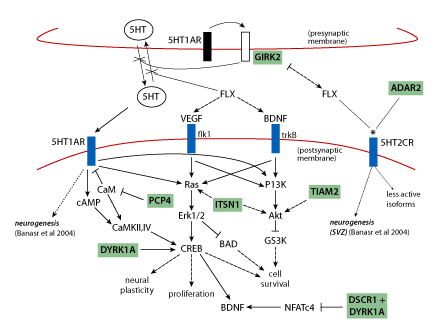
Figure 3 | Pathways relevant to adult neurogenesis: predicting perturbations in Down syndrome and effects of fluoxetine. Signalling through 5HT1AR is required for neurogenesis; VEGF and BDNF are required for proliferation of neural precursors and survival of newborn neurons, respectively. Serotonin (5HT) levels are decreased in Down syndrome, possibly due to GIRK2 activation by 5HT1A autoreceptors causing hyperpolarisation of the presynaptic membrane and inhibition of 5HT release. Fluoxetine (FLX) blocks reuptake of 5HT and thus increases 5HT concentrations and resultant signaling through the integrated pathways that involve cAMP, Ras/Erk and PI3K/Akt, and that converge on CREB. Several chromosome 21 proteins (shaded) interact with pathway components and are predicted to affect pathway flux when overexpressed. Fluoxetine also alters functional properties of 5HT2CR (*) which may also be affected by increased ADAR2(*). Solid arrows, activation; dashed arrows, indirect activation; blunt lines, inhibition; dotted lines, alters functional properties. (Figure is a composite from Warner-Schmidt and Duman, Banasr et al. and Gardiner et al. [25,26,27] ). With the exception of ADAR2, all chromosome 21 proteins are trisomic in the Ts65Dn mouse.
It is important, however, to keep in mind that the data in Table 1 represent the results of basic research, and while they are encouraging for their potential for application in Down syndrome, they are not clinical data. Each intervention has significant limitations that require resolution before clinical application in Down syndrome is considered. Applicable to all entries in Table 1 are the following: none has been approved for use in children, the consequences of long term use are not known (i.e. side effects, loss of efficacy), the extent of learning/memory benefits are not known (i.e. how many and which types of deficits can be rescued), and, above all, because the Ts65Dn is not a perfect model for Down syndrome, the effects on drug responses of the additional genes that are trisomic in Down syndrome are not known and could be both significant and negative.
Prediction of critical genes and potential therapeutic targets
Recent advances in genetics and biochemistry have made it increasingly clear that chromosome 21 proteins do not function independently. As shown in Figures 2 and 3, many have been shown to impact the same or overlapping biochemical pathways and cellular processes or share the same targets, and may act synergistically or antagonistically. There are a number of additional drugs/small molecules that could be used immediately in similar studies with 'knowledge-based' targets: TIAM1/Rac, Dyrk1a, MAPK, calcineurin, NMDAR, GABRA, neurogenesis (Figure 2, large arrows). These could be tested relatively quickly in learning/memory tasks in the Ts65Dn, and results would strengthen or refute the molecular basis of results in Table 1 and predictions in Figures 2 and 3. Verification of the learning/memory outcomes of several target-drug combinations would provide needed confidence for the potential benefits of preclinical assessments and initial clinical trials.
If a goal of Down syndrome research is knowledge-based identification of targets and potential pharmacotherapeutics, an aggressive programme should include a broad ranging survey for candidate chr21 genes and their connections to non-chromosome 21 genes with known functions in learning and memory processes. In Table 2, we list a number of Down syndrome features, seen with variable frequency and severity in the Down syndrome population, that directly or indirectly impact learning and memory, and also list candidate genes. Genes were chosen because, in each case, mutations have been identified that directly cause the specific feature. In Down syndrome, of course, we do not look for mutations, but the rationale is as follows: if a gene-specific mutation results, for example, in deafness, that gene must function in a pathway relevant to some aspect of the hearing process or development. Therefore it is conceivable that overexpression, as in Down syndrome, of some or all variants of the same gene may perturb the same pathway with deleterious consequences for hearing.
| Feature | Frequency in Down syndrome | hromosome 21 gene associations | Non-chromosome 21 gene associations |
|---|---|---|---|
| Cognitive deficits | Normal distribution; mean IQ = 45; range, 25-70 | RCAN1, ITSN1, SYNJ1, DYRK1A, TIAM1, PCP4, BACH1, SOD1, APP; microRNAs; *S100B; SIM2; DSCAM | NMDAR, MAPK, calcineurin, Ras, Elk, BDNF, GABRA; + 300 gene mutations associated with cognitive deficits |
| Memantine response | Unknown* | RCAN1, ITSN1, SYNJ1, DYRK1A, TIAM1, PCP4, BACH1, SOD1, APP | NR1, NR2a,b, calcineurin, calmodulin, GSK3B, Akt, Erk1/2 |
| Autism | 7% | CXADR-BTG3-NCAM2 region | NLGN3, NLGN4X; GABRA4; NRXN1(?) |
| Seizures | 7% | *CSTB, GRIK1 (JAE), GIRK2, HLCS, | +401 gene mutations associated with seizures |
| Hearing impairment | 15% | CLDN14 (DFNB29), *TMPRSS3 (DNFB8,10), KCNE1 (JLNS1) | +277 gene mutations associated with deafness |
| Vision impairment | 40% | *CRYAA, *CBS,* LSS | +155 gene mutations associated with vision impairment |
| Alzheimer's pathology | All | APP, BACE2 | APPL1, APPL2, BACE1, PS1, PS2, APOE, MAPT |
| AD-like dementia | 50% at age 50 | APP, BACE2 | APPL1, APPL2, BACE1, PS1, PS2, APOE, MAPT |
| Anxiety | ? | *SUMO3, *NRIP1; MRAP | GR, BDNF; MC2R |
| Early menopause | ? | *SUMO3, *NRIP1 | ER |
| Inflammation | ? | IFNAR1,2, IL10RB, IFNGR2,* S100B | IL1 |
| Down syndrome features and frequency [29-33]. Chromosome 21 and non-chromosome 21: Online Mendelian Inheritance in Man (https://www.ncbi.nlm.nih.gov/sites/entrez?db=omim). *, genes that are not present in the Ts65Dn model and thus their study requires development of new mouse models. | |||
Table 2 | Down syndrome features and variable frequency and candidate gene associations
We include non-chromosome 21 genes for a similar rationale: one or more specific variants of these genes, in the context of trisomy 21, may perturb normal functioning of the same pathways.
Forty chromosome 21 genes are included in Table 2. Not all of these can be studied in mice using the Ts65Dn and therefore additional mouse models need to be constructed. Table 2 also includes hundreds of non-chromosome 21 genes and it is likely that additional genes also contribute. While this seems a daunting number, there are large scale molecular approaches, such as microarray based procedures, that are now well established for handling >20,000 genes and even entire genome surveys. There is also the more novel but increasingly robust technique of high throughput DNA sequencing.
Large scale experiments may best be done with human DNA and this will require samples from individuals with Down syndrome. To be effective, this means the establishment of a bank of Down syndrome-derived DNAs, preferably as immortalised cell lines obtained from blood samples, plus the linking of each sample to a consistently detailed description of the individual's cognitive strengths and weaknesses. DNA banks, whole genome surveys and construction of new mouse models require a multi-year commitment of resources, including funding, and a realistic view of the time frame for translating successful basic research findings to clinical treatments.
Conclusions
Preventing or ameliorating the cognitive deficits in Down syndrome remains a challenge. However, recent successes with mouse models, increasing information on the functions and interactions of chromosome 21 genes, and increasingly sophisticated and high throughput technologies for assays of gene function suggest that an aggressive programme to identify and test potential pharmacotherapeutics can be productive. While use of mouse has practical advantages, it is important to be realistic about the limitations: there are significant and poorly understood differences in gene makeup and biochemistry between human and mouse that may affect the efficacy of any treatment; the costs of an appropriately aggressive research programme are significant, and the time and costs of clinical trials are considerably more so, especially when approval is sought for a new drug. However, while outcomes are not guaranteed, the potential is high for significant benefit to individuals with Down syndrome, and their families and communities. This potential should be explored.
References
- Huang X, Gardiner K. Protein coding features and conservation of novel human chr21 transcript, in preparation.
- Gardiner K, Costa AC. The genes of human chr21: choosing candidates for relevance to intellectual disability. American Journal of Medical Genetics. 2006;142:196-205.
- Gardiner K, Fortna A, Bechtel L and Davisson M. Mouse models of Down syndrome: how useful can they be? Comparison of the gene content of human chromosome 21 with orthologous mouse genomic regions. GENE. 2003;318:137-147.
- Mao R, Zielke CL, Zielke HR, Pevsner J. Global up-regulation of chr21 gene expression in the developing Down syndrome brain. Genomics. 2003;81:457-67.
- Kahlem P, Sultan M, Herwig R, Steinfath M, Balzereit D, Eppens B, Saran NG, Pletcher MT, South ST, Stetten G, Lehrach H, Reeves RH, Yaspo ML. Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of Down syndrome. Genome Research. 2004;14:1258-67.
- Gardiner K. Gene-dosage effects in Down syndrome and trisomic mouse models. Genome Biology. 2004;5:244.
- Dauphinot L, Lyle R, Rivals I, Dang MT, Moldrich RX, Golfier G, Ettwiller L, Toyama K, Rossier J, Personnaz L, Antonarakis SE, Epstein CJ, Sinet PM, Potier MC. The cerebellar transcriptome during postnatal development of the Ts1Cje mouse, a segmental trisomy model for Down syndrome. Human Molecular Genetics. 2005;14:373-84.
- Sultan M, Piccini I, Balzereit D, Herwig R, Saran NG, Lehrach H, Reeves RH, Yaspo ML. Gene expression variation in 'Down syndrome' mice allows to prioritize candidate genes. Genome Biology. 2007;8:R91.
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nature Genetics. 1995;1:177-184.
- Seregaza Z, Roubertoux PL, Jamon M, Soumireu-Mourat B. Mouse models of cognitive disorders in trisomy 21: a review. Behavior Genetics. 2006;36:387-404.
- O'Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, Cooke S, Sesay A, Modino S, Vanes L, Hernandez D, Linehan JM, Sharpe PT, Brandner S, Bliss TV, Henderson DJ, Nizetic D, Tybulewicz VL, Fisher EM. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science. 2005;309:2033-7.
- Li Z, Yu T, Morishima M, Pao A, Laduca J, Conroy J, Nowak N, Matsui S, Shiraishi I, Yu YE. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Human Molecular Genetics. 2007;16:1359-66.
- Lambert JC, Ferreira S, Gussekloo J, Christiansen L, Brysbaert G, Slagboom E, Cottel D, Petit T, Hauw JJ, DeKosky ST, Richard F, Berr C, Lendon C, Kamboh MI, Mann D, Christensen K, Westendorp R, Amouyel P. Evidence for the association of the S100beta gene with low cognitive performance and dementia in the elderly. Molecular Psychiatry. 2007;12:870-80.
- Pennington BF, Moon J, Edgin J, Stedron J, Nadel L. The neuropsychology of Down syndrome: evidence for hippocampal dysfunction. Child Development. 2003;74:75-93.
- Nade l, L. Down's syndrome: a genetic disorder in biobehavioral perspective. Genes, Brain and Behavior. 2003;2:156-166.
- Fernandez F, Morishita W, Zuniga E, Nguyen J, Blank M, Malenka RC, Garner CC. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nature Neuroscience. 2007;10:411-3.
- Costa AC, Scott-McKean JJ, Stasko MR. Acute Injections of the NMDA Receptor Antagonist Memantine Rescue Performance Deficits of the Ts65Dn Mouse Model of Down Syndrome on a Fear Conditioning Test. Neuropsycopharmacology. 2008;33:1624-32. [Epub 2007 Aug 15.]
- Rueda N, Flórez J, Martínez-Cué C. Chronic pentylenetetrazole but not donepezil treatment rescues spatial cognition in Ts65Dn mice, a model for Down syndrome. Neuroscience Letters. 2008;433:22-7.
- Clark S, Schwalbe J, Stasko MR, Yarowsky PJ, Costa ACS. Fluoxetine rescues deficient neurogenesis in hippocampus of the mouse model for Down syndrome Ts65Dn. Experimental Neurology. 2006:256-261.
- Cooper JD, Salehi A, Delcroix J-D, Hower CL, Belichenko PV, Cua-Couzens J, Kilbridge JR, Carlson EJ, Epstein CJ, Mobley WC. Failed retrograde transport of NGF in a mouse model of Down's syndrome: Reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10439-14444.
- Roper RJ, Baxter LL, Saran NG, Klinedinst DK, Beachy PA, Reeves RH. Defective cerebellar response to mitogenic Hedgehog signaling in Down [corrected] syndrome mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1452-6.
- Cataldo AM, Petanceska S, Peterhoff CM, Terio NB, Epstein CJ, Villar A, Carlson EJ, Staufenbiel M, Nixon RA. App gene dosage modulates endosomal abnormalities of Alzheimer's disease in a segmental trisomy 16 mouse model of down syndrome. Journal of Neuroscience. 2003;23:6788-6792.
- Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, Xia W, Villar A, Campbell WA, Kulnane LS, Nixon RA, Lamb BT, Epstein CJ, Stokin GB, Goldstein LS, Mobley WC. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29-42.
- Rueda N, Mostany R, Pazos A, Florez J, Martinez-Cue C. Cell proliferation is reduced in the dentate gyrus of aged but not young Ts65Dn mice, a model of Down syndrome. Neuroscience Letters. 2005;380:197-201.
- Warner-Schmidt JL, Duman RS. Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus. 2006;16:239-49.
- Banasr M, Hery M, Printemps R, Daszuta A. Serotonin-induced increases in adult cell proliferation and neurogenesis are mediated through different and common 5-HT receptor subtypes in the dentate gyrus and the subventricular zone. Neuropsychopharmacology. 2004;29:450-60.
- Gardiner K, Davisson MT, Crnic LS. Building protein interaction maps for Down syndrome. Briefings in Functional Genomics and Proteomics. 2004; 3:142-156.
- Wang J, Liu S, Haditsch U, Tu W, Cochrane K, Ahmadian G, Tran L, Paw J, Wang Y, Mansuy I, Salter MM, Lu YM. Interaction of calcineurin and type-A GABA receptor gamma 2 subunits produces long-term depression at CA1 inhibitory synapses. Journal of Neuroscience. 2003;23:826-36.
- Castillo H, Patterson B, Hickey F, Kinsman A, Howard JM, Mitchell T, Molloy CA. Difference in age at regression in children with autism with and without Down syndrome. Journal of Developmental and Behavioral Pediatrics. 2008;29:89-93.
- Cohen WI. Current dilemmas in Down syndrome clinical care: celiac disease, thyroid disorders, and atlanto-axial instability. American Journal of Medical Genetics Part C Seminar in Medical Genetics. 2006;142C:141-8.
- Dykens EM. Psychiatric and behavioral disorders in persons with Down syndrome. Mental Retardation and Developmental Disabilities Research Reviews. 2007;13:272-8.
- Shott SR. Down syndrome: common otolaryngologic manifestations. American Journal of Medical Genetics Part C Seminar in Medical Genetics. 2006;142C:131-40.
- Stephen E, Dickson J, Kindley AD, Scott CC, Charleton PM. Surveillance of vision and ocular disorders in children with Down syndrome. Developmental Medicine and Child Neurology. 2007;49:513-5.
Katheleen Gardiner is at the Department of Pediatrics, Human Medical Genetics Program, University of Colorado, Denver, 12800 E. 19th Avenue, Aurora, Colorado 80045, USA.
Correspondence to Katheleen Gardiner • e-mail: katheleen.gardiner@ucdenver.edu
Paper prepared from presentations and discussions at the Down Syndrome Research Directions Symposium 2007, Portsmouth, UK. The symposium was hosted by Down Syndrome Education International in association with the Anna and John J Sie Foundation, Denver. Major sponsors also included the Down Syndrome Foundation of Orange County, California and the National Down Syndrome Society of the USA. Information about the symposium can be found at https://www.dseinternational.org/research-directions /
doi:10.3104/reviews.2096
Received: 2 August 2008; Accepted: 9 August 2008; Published online: 24 October 2008

